Of the three, proteins (polypeptide chains) were the first to be sequenced.
The very earliest nucleic acid sequences were of RNA molecules. Robert Holley sequenced an alanine transfer RNA, 88 bases in length, and received the Nobel prize for this work in 1968 (pdf of Nobel lecture). The problem with tRNA is complicated by the presence of modified bases such as pseudouridine.

(There is a small problem with the structure shown in his lecture. This is yeast tRNA Phe, which is charged with phenylalanine).
There were other early pioneer chemists as well. Fred Sanger received two Nobel prizes, one for determining the amino acid sequence of insulin, and a second for developing the dideoxy sequencing method for DNA. He also worked on RNA sequencing methods.
RNA sequencing
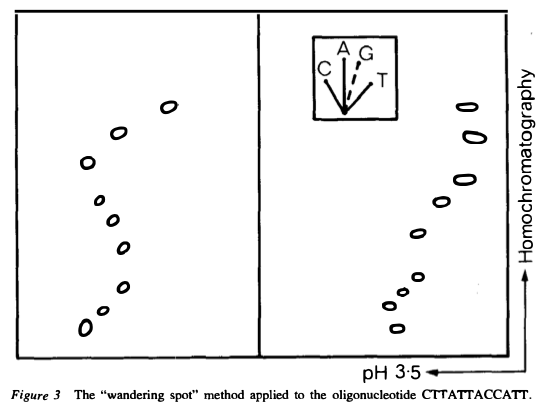
Some basic tools they used were: 32-P labeling, various sequence-specific nucleases including ribonuclease T1, and thin-layer chromatography and paper electrophoresis.
Labeling: The 32-P (pronounced P 32) isotope of phosphorous is radioactive (and energetic, emitting 1.7 MeV beta particles). Since DNA and RNA have phosphodiester bonds in them they can be made radioactive, incorporating 32-P by various means.
These include: growing cells in radioactive phosphate, (ii) using radioactive precursors for in vitro synthesis, and (iii) using the enzyme T4 polynucleotide kinase to add 32-P and label the 5' ends of molecules.
With a purified RNA, sequence analysis would usually start by making digests such as with T1 ribonuclease: which cuts specifically after G. So if an molecule was
pApCpApGpUpUpGpCpApApApUpCpA
T1 nuclease digestion would give three products
pApCpApGp
UpUpGp
CpApApApUpCpA
which can be individually purified. The sequences of the small fragments were then determined by a variety of methods. A different method was used to produce small fragments that overlap the joints.
Here are a couple of figures from Sanger's autobiographical Annual Reviews article which I hope convey the flavor of that kind of analysis, if not all the detail. The first shows a total digest of 5S rRNA.

A powerful idea is partial hydrolysis of end-labeled material.
lac operon control region
In 1975 the first natural DNA sequence was determined.

This was also the summer of the famous moratorium on recombinant DNA work. It was obvious that a new era was beginning for biology.
Reznikoff et al. used a genetic trick in E. coli to transfer the lac operon's promoter/operator region onto lambda phage, transcribed across the control region in vitro, and then used a biochemical trick to enrich for sequences that were only from the lac control region. The latter was to hybridize the RNA to separated strands of lambda phage without lac, and keep the stuff that didn't hybridize.
(PMID 1088926 --- this paper is locked behind a paywall at the journal Science, 45 years later, but I found a pdf linked on Bill Reznikoff's page at UW-Madison).
Maxam-Gilbert: chemical method
In 1977 Wally Gilbert's lab introduced a chemical sequencing method for DNA. I employed this method to sequence 70 bp of the promoter region for a late gene from phage T4 called gene 23. I used to boast that mine was the shortest DNA sequence ever published but that's not actually true. This one probably is.
The first step was to use alkaline phosphatase to remove 5' phosphates, then DNA kinase and 1 mCi of 32-P-ATP to label the ends. If you do this with double-stranded DNA then both ends will be labeled. You must somehow separate the strands or, cut with a restriction enzyme to get two different-sized pieces, and separate them by gel electrophoresis. Here's a strand separation from the paper.

The next steps were relatively simple organic chemistry
... the purines (A+G) are depurinated using formic acid, the guanines (and to some extent the adenines) are methylated by dimethyl sulfate [destabilizing the glycosidic bond], and the pyrimidines (C+T) are hydrolysed using hydrazine. The addition of salt (sodium chloride) to the hydrazine reaction inhibits the reaction of thymine for the C-only reaction. The modified DNAs may then be cleaved by hot piperidine; (CH2)5NH at the position of the modified base.What this does is to generate for each reaction a population of molecules, some of which end at the base in question. So if the original molecule is
*NpNpApNpNpNpApNpNpNpApN
with * marking the 32-P, then after partial cleavage after A (adenosine) you have
*NpNpApNpNpNpApNpNpNpApN
*NpNpApNpNpNpApNpNpNpA
*NpNpApNpNpNpA
*NpNpA
Here is the wikipedia figure:

Here is the wikipedia figure:

After gel electrophoresis and autoradiography (exposure of the gel to X-ray film), you get something like this:

And you can read the sequence.
Sanger sequencing
Fred Sanger developed a method for DNA sequencing. It generates a population of molecules like above by using synthesis instead of degradation. The synthesis is stopped short in some of the molecules by including a reagent which poisons the reaction, namely, a dideoxynucleotide triphosphate.

Here is the principle of the method in a figure from Sanger's (second) Nobel lecture.

In 1988, I spent about 3 months running about 100 sequencing gels to obtain about 3.5 kb of sequence data to determine the sequence of my favorite gene, hemA. The data looked like this:

With somewhat better methods I spent about 6 months to obtain about 12 kb of sequence data in 1993-94.
Year Amt Time invested
1983 70 bp months
1988 3500 bp 3 months
1994 12000 bp 6 months
Probably the biggest challenge in the method was to prepare the large polyacrylamide gels with no bubbles and a thickness at the top of 0.4 mm.
This era ended when commercial DNA sequencing services started. Together with PCR, including PCR to amplify transposon-genome junctions, it revolutionized our work in the mid-1990s. We would do a PCR reaction, send it in the mail, and 2 days later get a result by email.
Two very important adaptations were later made to the method. First, rather than label the DNA, the dideoxy terminators were themselves labeled with fluorescent dyes. Among other things, this meant that the reactions could be analyzed together in a single sample and analyzed by laser activation and a detector.

The other was replacement of polyacrylamide gels by capillary electrophoresis. This extended the amount of information obtainable on one analysis of a sample from about 200 nt to about 1000 nt.
Machines do many reactions in parallel. ABI 310. You can buy a used one cheap, but caveat emptor.
That is the technology which was used to sequence the human genome, declared complete in 2003.